

-
DFT-Based Studies on the Jahn-Teller Effect in 3d Hexacyanometalates with Orbitally Degenerate Ground States
M. Atanasov, P. Comba, C.A. Daul and A. Hauser
Journal of Physical Chemistry A, 111 (37) (2007), p9145-9163


DOI:10.1021/jp0731912 | unige:3584 | Abstract | Article HTML | Article PDF
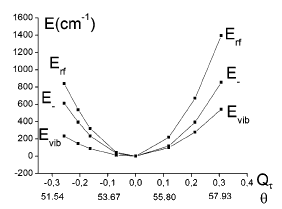
The topology of the ground-state potential energy surface of M(CN)6 with orbitally degenerate 2T2g (M = TiIII (t2g1), FeIII and MnII (both low-spin t2g5)) and 3T1g ground states (M = VIII (t2g2), MnIII and CrII (both low-spin t2g4)) has been studied with linear and quadratic JahnâTeller coupling models in the five-dimensional space of the εg and Ï2g octahedral vibrations (Tgâ(εg+Ï2g) JahnâTeller coupling problem (Tg = 2T2g, 3T1g)). A procedure is proposed to give access to all vibronic coupling parameters from geometry optimization with density functional theory (DFT) and the energies of a restricted number of Slater determinants, derived from electron replacements within the t2g1,5 or t2g2,4 ground-state electronic configurations. The results show that coupling to the Ï2g bending mode is dominant and leads to a stabilization of D3d structures (absolute minima on the ground-state potential energy surface) for all complexes considered, except for [Ti(CN)6]3-, where the minimum is of D4h symmetry. The JahnâTeller stabilization energies for the D3d minima are found to increase in the order of increasing CNâM Ï back-donation (TiIII < VIII < MnIII < FeIII < MnII < CrII). With the angular overlap model and bonding parameters derived from angular distortions, which correspond to the stable D3d minima, the effect of configuration interaction and spinâorbit coupling on the ground-state potential energy surface is explored. This approach is used to correlate JahnâTeller distortion parameters with structures from X-ray diffraction data. JahnâTeller coupling to trigonal modes is also used to reinterpret the anisotropy of magnetic susceptibilities and g tensors of [Fe(CN)6]3-, and the 3T1g ground-state splitting of [Mn(CN)6]3-, deduced from near-IR spectra. The implications of the pseudo JahnâTeller coupling due to t2gâeg orbital mixing via the trigonal modes (Ï2g) and the effect of the dynamic JahnâTeller coupling on the magnetic susceptibilities and g tensors of [Fe(CN)6]3- are also addressed.
